
RESEARCH ARTICLE
Regulation of Biogenesis and Fusion/Fission Processes of Vascular Mitochondria In Aldosterone-Induced Hypertension
Elena Olivares-Álvaro, María Belén Ruiz-Roso, Mercedes Klett-Mingo, Sandra Ballesteros, Ricardo Gredilla, Adrián Galiana-Simal, Natalia de las Heras, Vicente Lahera, Beatriz Martín-Fernández*
Article Information

Identifiers and Pagination:
Year: 2018Volume: 10
First Page: 76
Last Page: 85
Publisher Id: TOHYPERJ-10-76
DOI: 10.2174/1876526201810010076
Article History:
Received Date: 29/8/2018Revision Received Date: 15/10/2018
Acceptance Date: 31/10/2018
Electronic publication date: 28/12 /2018
Collection year: 2018
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: (https://creativecommons.org/licenses/by/4.0/legalcode). This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background:
Aldosterone plays a key role in the development of endothelial dysfunction and hypertension. The regulation of biogenesis and fusion/fission processes of vascular mitochondria has not been examined in aldosterone-induced hypertension. Thereby, we sought to explore in greater depth the role of aldosterone in mitochondrial biogenesis and fusion/fission processes in hypertension and the associated increases in oxidative stress.
Methods:
Male Wistar rats received aldosterone (1mg/Kg/day) + 1% NaCl as drinking water for 3 weeks.
Results:
Systolic blood pressure was elevated (p<0.05) in aldosterone-treated rats. eNOS and p-eNOSSer1177 protein expression was down regulated (p<0.05) and NADPH oxidase subunit p22phox expression was increased (p<0.05) in aldosterone-treated rats. Expression of mitochondrial biogenesis proteins SIRT1, PGC1α, PPARγ, and TFAM decreased (p<0.05) in aldosterone-treated rats. Protein expression of vascular DRP1, OMA1 and S-OPA1 up regulated (p<0.05) in aldosterone-treated rats. MFN1 and L-OPA1 (p<0.05) decreased in aldosterone-treated animals.
Conclusion:
The results showed that, in aldosterone-treated rats, hypertension is likely associated with increased oxidative stress in the aorta and with changes in the regulation of two key mitochondrial processes such as biogenesis and fusion/fission processes. The overall mitochondrial alterations observed in the study may play a role in aldosterone-derived vascular oxidative stress and hypertension.
1. INTRODUCTION
Aldosterone mediates endothelial dysfunction and promotes hypertension [1-4]. Blockade of Mineralocorticoid Receptors (MR) prevents endothelial nitric oxide synthase (eNOS) downregulation associated with hypertension and heart failure in experimental studies [5-7]. Thus, previous data support the participation of aldosterone in inhibiting the enzyme involved in NO synthesis in these pathological situations. In addition to vascular NADPH oxidase-derived ROS, a role of aldosterone in the progression of mitochondrial and cardiovascular dysfunction has been suggested [8, 9]. In cardiac experimental models, inhibition of the renin-angiotensin-aldosterone-system (RAAS) with either angiotensin II (Ang II) receptor and/or mineralocorticoid receptor blockade, attenuates the mitochondrial abnormalities observed [10, 11]. These findings suggest a role of mitochondrial changes in cardiovascular alterations associated with RAAS impairment.
Mitochondria are highly dynamic organelles involved in cell metabolism and different signaling pathways [12]. Mitochondrial biogenesis is driven, in part, through the peroxisome proliferator-activated receptor gamma (PPARγ) coactivator-1alpha (PGC1α). PGC1α has been shown to mediate downstream transcriptional regulatory circuits including nuclear respiratory factor-1 and 2 (NRF1 and NRF2). NRF1 regulates a number of mitochondrial genes such as cytochrome c oxidase subunit 5B (COX5B), Succinate Dehydrogenase complex iron-sulfur subunit B (SDHB) and mitochondrial transcription factor A (TFAM) (Klinge, 2008). Sirtuins play an important role in the regulation of both biogenesis and degradation of mitochondria. In particular, SIRT1 has been shown to regulate these processes under stress circumstances leading to decreased PGC1α expression in rats [13].
The mitochondrial network is constantly exposed to fusion/fission events. The “balance” between those processes leads to the formation of elongated or discrete fragmented mitochondria [14]. These processes are essential for maintaining an efficient mitochondrial network. Fusion events allow the mixing of intra-mitochondrial proteins and the replacement of damaged mitochondrial DNA [15]. In the outer mitochondrial membrane (OMM), two main proteins, mitofusins 1 (MFN1) and 2 (MFN2) mediate the fusion of the OMM, whereas in the inner mitochondrial membrane (IMM), optic atrophy 1 (OPA1) regulates the fusion of the IMM [16]. Under physiological conditions, proteolytic cleavage of long isoforms of OPA1 (L-OPA1) results in the balanced accumulation of long and short OPA1 (S-OPA1) forms [17-20]. On the other hand, proteolysis of OPA1 by the metalloendopeptidase OMA1 leads to the loss of the L-OPA1. OMA1 has been described as a new key mitochondrial regulator whichability of processing OPA1 is activated in response to some stress stimuli [21, 22]. OPA1 proteolysis by OMA1 results in the inactivation of OPA1 and diminished inner membrane fusion [17, 21, 23] and by hence, increased fission process. Regarding mitochondrial fission, it is mainly mediated by dynamin-related protein1 (DRP1) which translocates from the cytosol to the OMM where it interacts with other proteins of the fission machinery including human fission protein 1, mitochondrial fission factor, and mitochondrial dynamics proteins of 49 and 51 kDa [24, 25]. The involvement of mitochondrial alterations in the pathogenesis of hypertension has been associated with mitochondrial energy deficiency [26]. However, mitochondrial alterations not only affect bioenergetics, but also play a role in apoptotic events, which participate in the decline of tissue functionality [27].
When mitochondria become maladaptive, due to loss and/or reduced efficiency of mitochondria, a cascade of events develops including reduced mitochondrial biogenesis, altered fusion/fission processes and ROS production. As previously cited, a number of seminal studies have proposed a role for the RAAS and the associated increase in oxidative stress, in the mitochondrial alterations [8, 9, 28]. However, the regulation of biogenesis and fusion/fission processes of vascular mitochondria has not been investigated in aldosterone-induced hypertension. Thereby, we sought to explore in greater depth the role of aldosterone in mitochondrial biogenesis and fusion/fission processes in hypertension and the associated increase in oxidative stress.
2. METHODS
2.1. Experimental Design and Animal Model
The study was conducted in 20 male Wistar rats (250 g, Harlan, Horst, Holand). The Universidad Complutense Ethics Review Board specifically approved this study according to the guidelines for ethical care of experimental animals of the European Union and granted and approved by the Universidad Complutense Ethics Review Board following the National Guideline 53/2013. Rats were fed standard rat chow and tap water ad libitum and kept in a quiet room at constant temperature (20-22oC) and humidity (50-60%). Before allocating animals to treatment, systolic blood pressure (SBP) was measured to group them under the same mean SBP by the tail-cuff method [29, 30]. Rats were treated with either aldosterone (ALDO, 1mg/Kg/day, Sigma Aldrich, Germany) plus 1% NaCl as drinking water, or vehicle (CONTROL, sunflower oil), subcutaneously injected daily for 3 weeks.
2.2. Systolic Blood Pressure Measurements
SBP was measured by the tail-cuff method [29, 30] at the end of the treatment period with Niprem 645, Cibertec, Madrid, Spain. Average of 10 measurements were taken as final SBP.
2.3. Western Blot Analysis
Aorta protein samples (100mg) were homogenized in lysis buffer (50 mM TrisHCl, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.2% Triton X-100, 0.3% NP-40, 0.1 mM PMSF, and 1 mg/ml pepstatin A). Homogenates containing 40 µg protein were separated in a 10% SDS-PAGE under reducing conditions and then transferred to a polivinilifluoride membrane (Bio-rad). Membranes were blocked in 7.5% nonfat milk in PBS+0.1% Tween 20 (PBS-T). Antibodies to: mouse monoclonal anti-mitofusin 1 (1:500, ab57602, Abcam, UK), anti-DRP1 (1:500, ab56788, Abcam, UK), anti-SIRT1 (1:500, ab110304, Abcam, UK), and anti-OPA1 (1:1000, 612607, BD Biosciences, USA), rabbit polyclonal anti-eNOS (1:250, ab95254, Abcam, UK), anti-phospho-eNOSser1177 (1:1000, 9571, Cell signaling technology, USA), anti-p22phox (1:500, sc-20781, Santa Cruz Biotechnology, Germany), anti-TFAM (1:1000, ab131607, Abcam, UK), anti-NRF1 (1:500, ab175932, Abcam, UK), anti-OMA1 (1:500, ab104316, Abcam, UK), and anti-PPARγ (1:500, sc-7196, Santa Cruz Biotechnology, Germany), goat polyclonal anti-PGC1α (1:250, ab106814, Abcam, UK) were used. Immunoreactive proteins were detected by chemiluminescence with ECLPlus (Millipore-Bedford, Boston, USA). Blots were probed with rabbit monoclonal anti-B-actin (1:10000) (Sigma Aldrich, Co, Spain) as an internal control, to normalize between gels. Quantification was expressed as a percentage of relative protein expression (Protein/ B-actin) vs CONTROL group.
2.4. Statistical Analysis
All analyses and graphs were performed using GraphPad Prism 5 (GraphPad Software Inc., USA). Values are presented as the mean ± standard error of the mean (SEM). The data were compared by the Lilliefors test followed by Student´s T-test. Data are presented as means 10 SEMs, and the level of significance was set at p < 0.05.
3. RESULTS
3.1. Systolic Blood Pressure
As shown in Fig. (1), SBP was higher (p<0.05) in ALDO group than in CONTROL group (Fig. 1).
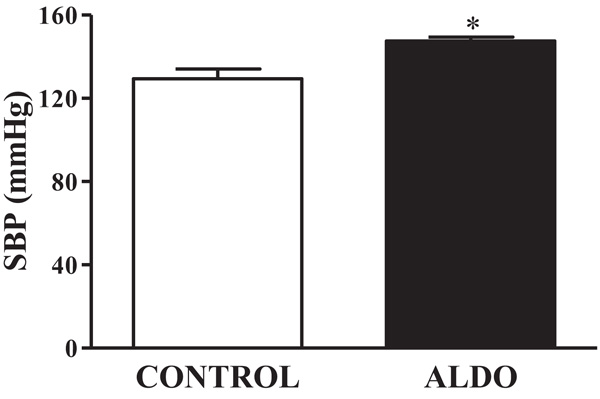 |
Fig. (1). Effect of aldosterone on SBP (mmHg) measured by tail-cuff method. Data are expressed as percentage of mean ± SEM versus CONTROL group. * p<0.05 vs. CONTROL. CONTROL: Control group; ALDO: Aldosterone-salt group; (n=20). |
3.2. Vascular eNOS and p-eNOS Ser1177
Both vascular protein expression of eNOS and p-eNOSSer1177 were decreased (p<0.05) in ALDO group when compared to CONTROL (Figs. 2A and 2B).
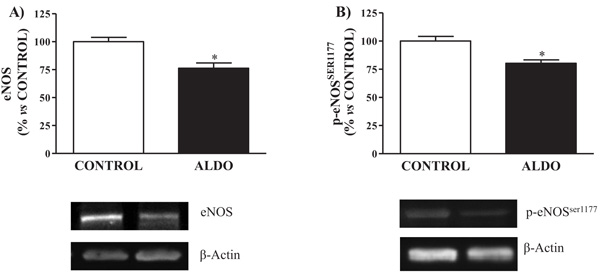 |
Fig. (2). Effect of aldosterone on aorta eNOS and p-eNOSSer1177 protein expression measured by Western Blot analysis. Relative protein expression of, A) endothelial nitric oxide synthase (eNOS) and B) endothelial nitric oxide synthase phosphorylation on Ser1177 (p-eNOSSer1177). Data are expressed as percentage of mean ± SEM versus CONTROL group. *p < 0.05 vs. CONTROL. CONTROL: Control group; ALDO: Aldosterone-salt group; (n=20). |
3.3. Vascular NADPH Oxidase-P22phox
Protein expression of vascular NADPH oxidase-derived ROS, p22phox, was upregulated (p<0.05) in ALDO rats when compared to CONTROL (Fig. 3).
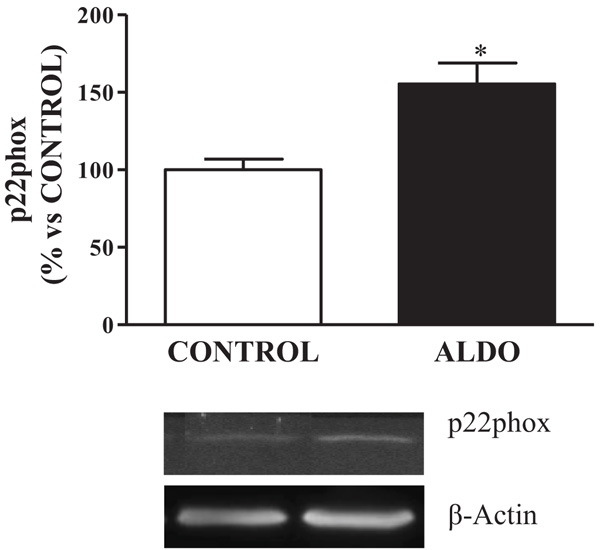 |
Fig. (3). Effect of aldosterone on oxidative stress. Relative protein expression of, A) NADPH oxidase subunit p22phox (p22phox). Data are expressed as percentage of mean ± SEM versus CONTROL group. *p < 0.05 vs. CONTROL. CONTROL: Control group; ALDO: Aldosterone-salt group; (n=20). |
3.4. Vascular Mitochondrial Biogenesis Proteins
Aldosterone-treated rats showed decreased (p<0.05) PGC1α protein expression when compared to CONTROL rats Fig. (4A). Mitochondrial biogenesis protein NRF1 tended to decrease in ALDO group while TFAM showed decreased (p<0.05) levels when compared with CONTROL Fig. (4B and C). Decreased (p<0.05) SIRT1 and PPARγ vascular protein expression were observed in the ALDO group rats compared to CONTROL (Figs. 4D and E).
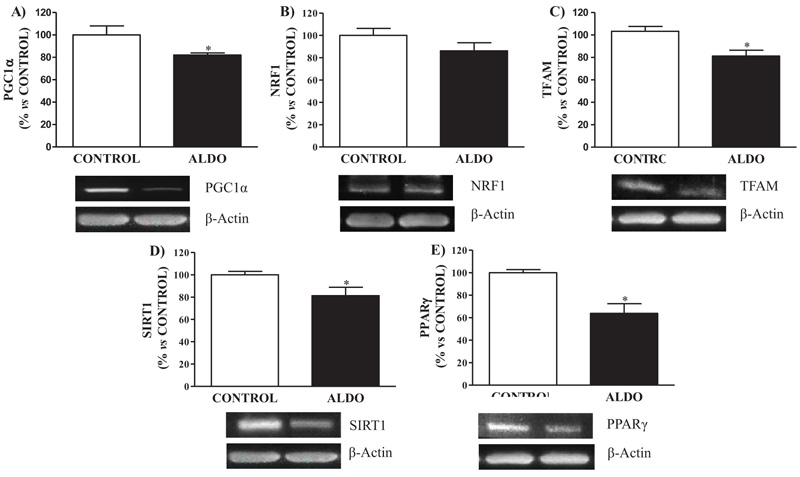 |
Fig. (4). Effect of aldosterone on vascular mitochondrial biogenesis. Relative protein expression measured by Western Blot analysis of, A) Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1a), B) Nuclear Respiratory Factor 1 (NRF1), C) mitochondrial transcription factor A (TFAM), D) Sirtuin 1 (SIRT1), E) Peroxisome proliferator-activated receptor gamma (PPARg). Data are expressed as percentage of mean ± SEM versus CONTROL group. *p < 0.05 vs. CONTROL. CONTROL: Control group; ALDO: Aldosterone-salt group; (n=20). |
3.5. Vascular Mitochondrial Fusion/Fission Processes Proteins
Mitochondrial fusion protein MFN1 was downregulated (p<0.05) in ALDO group when compared to CONTROL Fig. (5A). Decreased (p<0.05) levels of isoform L-OPA1 and increased (p<0.05) levels of S-OPA1 in aorta were observed in aldosterone-treated rats when compared to CONTROL Figs. (5B and D). Vascular protein expression of mitochondrial fission protein DRP1 was higher (p<0.05) in ALDO group than in the CONTROL group Fig. (5C). Metalloendopeptidase OMA1 protein expression in the aorta was higher (p<0.05) in ALDO group than in the CONTROL group (Fig. 5D).
4. DISCUSSION
The present results show that, in aldosterone-treated rats, hypertension is associated with changes in the regulation of biogenesis and fusion/fission processes of aortic mitochondria. Aldosterone altered mitochondrial biogenesis by decreasing PGC1α, NRF1, TFAM, SIRT1 and PPARγ protein expression. Likewise, aldosterone enhanced fission processes up-regulating OMA-1, decreasing L-OPA1 isoform and increasing S-OPA1 isoform protein expression. MFN1 and DRP1 protein expression were also altered in aldosterone-treated rats. The overall observed data suggest a role of aorta mitochondrial alterations in aldosterone-derived vascular oxidative stress and hypertension.
Aldosterone increases NADP(H] oxidase activity and oxidative stress in aorta through an MR-dependent mechanism [31, 32]. The NADPH oxidase is widely expressed in the vascular system. It is expressed in vascular smooth muscle cells, adventitial fibroblasts, endothelial cells and perivascular adipocytes [33-35]. It is well known that excess of aldosterone leads to an increase of NADPH oxidase activity and ROS production in the vascular wall. Specifically, p22phox sub-unity is widely expressed in the vascular wall and it has been demonstrated that high levels of aldosterone induce its overexpression in the cardiovascular system [36, 37]. The generation of ROS by aldosterone further leads to endothelial dysfunction decreasing eNOS which is associated with hypertension [2, 5, 7]. eNOS plays an important role in mitochondria biogenesis and mitochondrial function [9, 38, 39]. For instance, in the myocardium, NO has been shown to stimulate mitochondrial biogenesis [40]. Moreover, reductions in mitochondria content and associated defects in fatty acid metabolism are evident in eNOS-deficient mice that manifest insulin resistance and hypertension [39]. Reduced eNOS levels in myocardial dysfunction have been also related to PGC1α reduced expression, its downstream nuclear factors, and attenuation of mitochondrial biogenesis [41]. Ang II-mediated mitochondrial dysfunction in vascular endothelial cells has been previously described [8]. In the present study, aldosterone increased NADPH oxidase component p22phox and decreased eNOS and p-eNOSser1177 protein expression contributing to oxidative stress and hypertension. Our data provide the first evidence that, in the aorta, aldosterone-induced eNOS uncoupling is accompanied not only by reduced PGC1α levels but also downstream transcriptional regulatory mediator NRF1 and the oxidative phosphorylation mediator TFAM. These data collectively seems to underscore the important functional role of eNOS in the maintenance of vascular mitochondrial biogenesis and mitochondrial dynamic processes in elevated aldosterone situations.
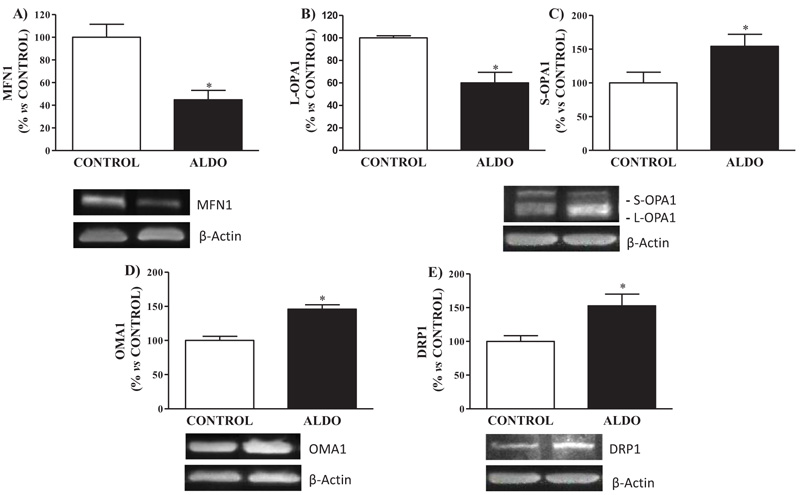 |
Fig. (5). Effect of aldosterone on vascular mitochondrial morphology. Relative protein expression measured by Western Blot analysis of, A) Mitofusin 1 (MFN1), B) Optic atrophy type 1 long-isoform (L-OPA1) C) Optic atrophy type 1 short-isoform (S-OPA1), D) Dynamin-related protein 1 (DRP1) and E) Mitochondrial metalloendopetidase (OMA1). Data are expressed as percentage of mean ± SEM versus CONTROL group. * p < 0.05 vs. CONTROL. CONTROL: Control group; ALDO: Aldosterone-salt group; (n=20). |
Moreover, we observed decreased SIRT1 and PPARγ vascular protein expression induced by aldosterone. SIRT1 has been identified as a modulator of the aldosterone signaling pathway [42, 43]. SIRT1 plays an essential role in the genesis of aldosterone-induced renal injury which is directly related to hypertension. One of the best-characterized targets of SIRT1 is PGC1α [44] since PGC1α activates NRF1 to promote mitochondrial biogenesis. It has been proposed that SIRT1 might be able to modulate the creation of new mitochondria during tissue repair in cases of cardiac injury [45]. By decreasing PGC1α vascular expression, aldosterone might modulate mitochondria biogenesis which may be playing a role in hypertension.
As previously mentioned, PGC1α is a key mediator of mitochondrial biogenesis and an inducer of MFN1 which promotes mitochondrial fusion of the OMM. Mitochondrial fusion and fission processes ensure equal division of mitochondrial numbers during cell division and mediate the selective removal of damaged mitochondria by the process of mitophagy while the fusion process is activated during conditions of increased mitochondrial bioenergetics [13]. The fission process is activated during mitochondrial degradation through the mitophagy [46]. Disrupted fusion and fission regulatory pathways exacerbate ROS production. For instance, Opa1 +/- mice displayed enlarged mitochondria and disrupted cristae leading to cardiomyopathy (Walters, et al. 2016). In our study, aldosterone compromised vascular mitochondrial fusion process decreasing MFN1 expression and inducing OPA1 expression, by hence, decreasing L-OPA1 isoform in detriment of S-OPA isoform. Alterations in the fusion/fission process derived from aldosterone administration might induce mitochondrial changes thus contributing to the higher oxidative stress observed in hypertension. Interestingly, protection of cardiac tissue by lowering mitochondrial metabolism can occur with DRP1 inhibition [47]. Data in our study also showed increased DRP1 protein expression in the aorta of aldosterone-treated rats contributing to the role of mitochondrial fusion/fission processes alteration to hypertension via oxidative stress.
CONCLUSION
In summary, our results suggest a significant role of vascular mitochondrial alterations in aldosterone-induced hypertensive status and the associated increased vascular oxidative stress. Reduced efficiency of aortic mitochondria seems to be developing a cascade of events including reduced mitochondrial biogenesis, altered fusion/fission processes, and ROS production, all of which may contribute to hypertension. The study provides interesting results which might be the starting point for further studies exploring the role of aldosterone in the mitochondrial biogenesis and fusion/fission process in hypertension.
FUNDING
This work was supported by grants from Ministerio de Economía y Competitividad Retos-Colaboración (RTC-2014-1689-1).
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The Universidad Complutense Ethics Review Board specifically approved this study.
HUMAN AND ANIMAL RIGHTS
The study has been approved according to the guidelines for ethical care of experimental animals of the European Union.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.
